
ГОСТ 25346-89: Основные нормы взаимозаменяемости. Единая система допусков и посадок. Общие положения, ряды допусков и основных отклонений
Терминология ГОСТ 25346-89: Основные нормы взаимозаменяемости. Единая система допусков и посадок. Общие положения, ряды допусков и основных отклонений оригинал документа:
1.1.19. Вал — термин, условно применяемый для обозначений наружных элементов деталей, включая и нецилиндрические элементы.
Определения термина из разных документов: Вал
1.1.10. Верхнее отклонение ES , es — алгебраическая разность между наибольшим предельным и соответствующим номинальным размерами (черт. 2).
Примечание. ES — верхнее отклонение отверстия; es — верхнее отклонение вала.
Определения термина из разных документов: Верхнее отклонение ES
1.1.8. Действительное отклонение — алгебраическая разность между действительным и соответствующим номинальным размерами.
Определения термина из разных документов: Действительное отклонение
1. 1.2. Действительный размер — размер элемента, установленный измерением.
1.2. Действительный размер — размер элемента, установленный измерением.
Определения термина из разных документов: Действительный размер
1.1.27. Допуск посадки — сумма допусков отверстия и вала, составляющих соединение.
Определения термина из разных документов: Допуск посадки
1.1.14. Допуск Т — разность между наибольшим и наименьшим предельными размерами или алгебраическая разность между верхним и нижним отклонениями (черт. 2).
Примечание. Допуск — это абсолютная величина без знака.
Определения термина из разных документов: Допуск Т
1.1.18. Единица допуска i, I — множитель в формулах допусков, являющийся функцией номинального размера и служащий для определения числового значения допуска.
Примечание. i — единица допуска для номинальных размеров до 500 мм, I — единица допуска для номинальных размеров св. 500 мм.
Определения термина из разных документов: Единица допуска
1.1.28. Зазор — разность между размерами отверстия и вала до сборки, если размер отверстия больше размера вала (черт. 3).
3).
Определения термина из разных документов: Зазор
1.4. Интерпретация предельных размеров
Для отверстий — диаметр наибольшего правильного воображаемого цилиндра, который может быть вписан в отверстие так, чтобы плотно контактировать с наиболее выступающими точками поверхности на длине соединения (размер сопрягаемой детали идеальной геометрической формы прилегающей к отверстию без зазора), не должен быть меньше, чем предел максимума материала. Дополнительно наибольший диаметр в любом месте отверстия, определенный путем двухточечного измерения, не должен быть больше, чем предел минимума материала.
Для валов — диаметр наименьшего правильного воображаемого цилиндра, который может быть описан вокруг вала так, чтобы плотно контактировать с наиболее выступающими точками поверхности на длине соединения (размер сопрягаемой детали идеальной геометрической формы прилегающей к валу без зазора), не должен быть больше чем предел максимума материала. Дополнительно наименьший диаметр в любом месте вала, определенный путем двухточечного измерения, не должен быть меньше, чем предел минимума материала.
Дополнительная информация к интерпретации предельных размеров приведена в приложении 2.
Определения термина из разных документов: Интерпретация предельных размеров
1.1.17. Квалитет (степень точности) — совокупность допусков, рассматриваемых как соответствующие одному уровню точности для всех номинальных размеров.
Определения термина из разных документов: Квалитет
1.1.34. Наибольший зазор — разность между наибольшим предельным размером отверстия и наименьшим предельным размером вала в посадке с зазором или в переходной посадке (черт. 8 и 9).
Определения термина из разных документов: Наибольший зазор
1.1.36. Наибольший натяг — разность между наибольшим предельным размером вала и наименьшим предельным размером отверстия до сборки в посадке с натягом или в переходной посадке (черт. 9 и 10).
Определения термина из разных документов: Наибольший натяг
1.1.4. Наибольший предельный размер — наибольший допустимый размер элемента (черт. 1).
1).
Определения термина из разных документов: Наибольший предельный размер
1.1.33. Наименьший зазор — разность между наименьшим предельным размером отверстия и наибольшим предельным размером вала в посадке с зазором (черт. 8).
Черт. 5 | Черт. 6 | Черт. 7 | |
Черт. 8 | Черт. 9 | ||
Черт. 10 | |||
Определения термина из разных документов: Наименьший зазор
1. 1.35. Наименьший натяг — разность между наименьшим предельным размером вала и наибольшим предельным размером отверстия до сборки в посадке с натягом (черт. 10).
1.35. Наименьший натяг — разность между наименьшим предельным размером вала и наибольшим предельным размером отверстия до сборки в посадке с натягом (черт. 10).
Определения термина из разных документов: Наименьший натяг
1.1.5. Наименьший предельный размер — наименьший допустимый размер элемента (черт. 1).
Определения термина из разных документов: Наименьший предельный размер
1.1.29. Натяг — разность между размерами вала и отверстия до сборки, если размер вала больше размера отверстия (черт. 4).
Примечание. Натяг можно определять как отрицательную разность между размерами отверстая и вала.
Определения термина из разных документов: Натяг
1.1.11. Нижнее отклонение EI , ei — алгебраическая разность между наименьшим предельным и соответствующим номинальным размерами (черт. 2).
Примечание. ЕI — нижнее отклонение отверстия; ei — нижнее отклонение вала.
Определения термина из разных документов: Нижнее отклонение EI
1. 1.6. Номинальный размер — размер, относительно которого определяются отклонения (черт. 1 и 2).
1.6. Номинальный размер — размер, относительно которого определяются отклонения (черт. 1 и 2).
Определения термина из разных документов: Номинальный размер
1.1.26. Номинальный размер посадки — номинальный размер, общий для отверстия и вала, составляющих соединение.
Определения термина из разных документов: Номинальный размер посадки
1.2. Нормальная температура
Допуски и предельные отклонения, установленные в настоящем стандарте, относятся к размерам деталей при температуре 20 °С.
Определения термина из разных документов: Нормальная температура
1.1.13. Нулевая линия — линия, соответствующая номинальному размеру, от которой откладываются отклонения размеров при графическом изображении полей допусков и посадок. Если нулевая линия расположена горизонтально, то положительные отклонения откладываются вверх от нее, а отрицательные — вниз (черт. 2).
Определения термина из разных документов: Нулевая линия
1.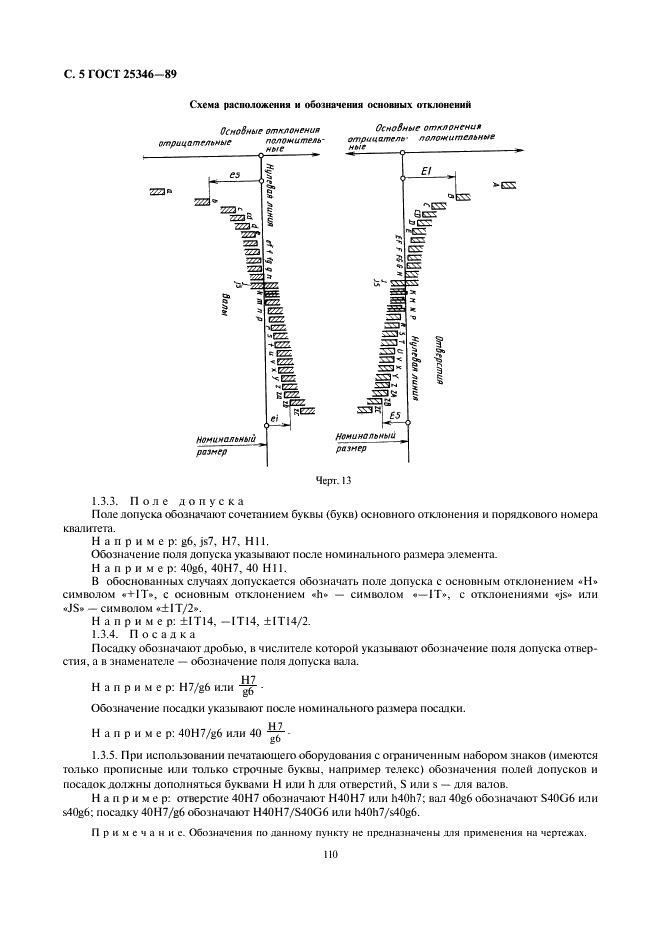 1.22. Основное отверстие — отверстие, нижнее отклонение которого равно нулю.
1.22. Основное отверстие — отверстие, нижнее отклонение которого равно нулю.
Черт. 3 | Черт. 4 |
Определения термина из разных документов: Основное отверстие
1.1.12. Основное отклонение — одно из двух предельных отклонений (верхнее или нижнее), определяющее положение поля допуска относительно нулевой линии. В данной системе допусков и посадок основным является отклонение, ближайшее к нулевой линии.
Черт. 1 | Черт. 2 |
Определения термина из разных документов: Основное отклонение
1.1.21. Основной вал — вал, верхнее отклонение которого равно нулю.
Определения термина из разных документов: Основной вал
1.1.20. Отверстие — термин, условно применяемый для обозначения внутренних элементов деталей, включая и нецилиндрические элементы.
Определения термина из разных документов: Отверстие
1.1.7. Отклонение — алгебраическая разность между размером (действительным или предельным размером) и соответствующим номинальным размером.
Определения термина из разных документов: Отклонение
1.1.32. Переходная посадка — посадка, при которой возможно получение как зазора, так и натяга в соединении, в зависимости от действительных размеров отверстия и вала. При графическом изображении поля допусков отверстия и вала перекрываются полностью или частично (черт. 7).
Определения термина из разных документов: Переходная посадка
1.1.16. Поле допуска — поле, ограниченное наибольшим и наименьшим предельными размерами и определяемое величиной допуска и его положением относительно номинального размера. При графическом изображении поле допуска заключено между двумя линиями, соответствующими верхнему и нижнему отклонениям относительно нулевой линии (черт. 2).
При графическом изображении поле допуска заключено между двумя линиями, соответствующими верхнему и нижнему отклонениям относительно нулевой линии (черт. 2).
Определения термина из разных документов: Поле допуска
1.1.25. Посадка — характер соединения двух деталей, определяемый разностью их размеров до сборки.
Определения термина из разных документов: Посадка
1.1.30. Посадка с зазором — посадка, при которой всегда образуется зазор в соединении, т.е. наименьший предельный размер отверстия больше наибольшего предельного размера вала или равен ему. При графическом изображении поле допуска отверстия расположено над полем допуска вала (черт. 5).
Определения термина из разных документов: Посадка с зазором
1.1.31. Посадка с натягом — посадка, при которой всегда образуется натяг в соединении, т.е. наибольший предельный размер отверстия меньше наименьшего предельного размера вала или равен ему. При графическом изображении поле допуска отверстия расположено под полем допуска вала (черт. 6).
6).
Определения термина из разных документов: Посадка с натягом
1.1.38. Посадки в системе вала
Определения термина из разных документов: Посадки в системе вала
1.1.37. Посадки в системе отверстия — посадки, в которых требуемые зазоры и натяги получаются сочетанием различных полей допусков валов с полем допуска основного отверстия (черт. 11).
Определения термина из разных документов: Посадки в системе отверстия
1.1.23. Предел максимума материала — термин, относящийся к тому из предельных размеров, которому соответствует наибольший объем материала, т.е. наибольшему предельному размеру вала или наименьшему предельному размеру отверстия.
Примечание. Применявшийся ранее термин «проходной предел» использовать не рекомендуется.
Определения термина из разных документов: Предел максимума материала
1. 1.24. Предел минимума материала — термин, относящийся к тому из предельных размеров, которому соответствует наименьший объем материала, т.е. наименьшему предельному размеру вала или наибольшему предельному размеру отверстия.
1.24. Предел минимума материала — термин, относящийся к тому из предельных размеров, которому соответствует наименьший объем материала, т.е. наименьшему предельному размеру вала или наибольшему предельному размеру отверстия.
Примечание. Применявшийся ранее термин «непроходной предел» использовать не рекомендуется.
Определения термина из разных документов: Предел минимума материала
1.1.9. Предельное отклонение — алгебраическая разность между предельным и соответствующим номинальным размерами. Различают верхнее и нижнее предельные отклонения.
Определения термина из разных документов: Предельное отклонение
1.1.3. Предельные размеры — два предельно допустимых размера элемента, между которыми должен находиться (или которым может быть равен) действительный размер (см. п. 1.4).
Определения термина из разных документов: Предельные размеры
1.1.1. Размер — числовое значение линейной величины (диаметра, длины и т.п.) в выбранных единицах измерения.
Определения термина из разных документов: Размер
1.1.15. Стандартный допуск IT — любой из допусков, устанавливаемых данной системой допусков и посадок.
Примечание. В дальнейшем в стандарте под термином «допуск» понимается «стандартный допуск».
Определения термина из разных документов: Стандартный допуск IT
1.3. Условные обозначения
1.3.1. Квалитеты
Квалитеты обозначаются порядковыми номерами, например, 01, 7, 14.
Допуски по квалитетам обозначаются сочетанием прописных букв IT с порядковым номером квалитета, например, IТ01, IТ7, IТ14.
Черт. 11 | Черт. 12 |
Определения термина из разных документов: Условные обозначения
Словарь-справочник терминов нормативно-технической документации. academic.ru.
2015.
academic.ru.
2015.
ГОСТ 25347-2013 (ISO 286-2:2010). Основные нормы взаимозаменяемости. Характеристики изделий геометрические. Система допусков на линейные размеры. Ряды допусков, предельные отклонения отверстий и валов
| Вид документа | ГОСТ |
| Статус | Действует |
| Документ принят организацией | Межгосударственный Совет по стандартизации метрологии и сертификации |
| Документ внесен организацией | Федеральное агентство по техническому регулированию и метрологии (Росстандарт) |
| Разработчик документа | Открытое акционерное общество «Научно-исследовательский и конструкторский институт средств измерений в машиностроении» (ОАО «НИИизмерения») |
| Дата принятия в МГС | 2013-11-14 |
| Дата начала действия | 2015-07-01 |
| Дата последней редакции | 2014-10-02 |
| Страны действия | Республика Беларусь;Республика Казахстан;Кыргызская Республика;Российская Федерация;Республика Таджикистан;Украина |
| Где применяется | Настоящий стандарт содержит числовые значения предельных отклонений отверстий и валов для классов допусков общего применения, вычисленные по ГОСТ 25346. В настоящем стандарте приведены значения верхних предельных отклонений отверстий ES и валов es, а также нижних предельных отклонений отверстий EI и валов ei. Система допусков ИСО на линейные размеры распространяется на следующие геометрические элементы: а) цилиндр; b) две параллельные противолежащие плоскости. В настоящем стандарте с целью упрощения подробно рассмотрены только детали представляющие собой круговые цилиндры, т.к. такие детали имеют важное значение. Однако допуски и отклонения, приведенные в настоящем стандарте, также могут относиться и к деталям, сечение которых не является окружностью В настоящем стандарте приведены значения верхних предельных отклонений отверстий ES и валов es, а также нижних предельных отклонений отверстий EI и валов ei. Система допусков ИСО на линейные размеры распространяется на следующие геометрические элементы: а) цилиндр; b) две параллельные противолежащие плоскости. В настоящем стандарте с целью упрощения подробно рассмотрены только детали представляющие собой круговые цилиндры, т.к. такие детали имеют важное значение. Однако допуски и отклонения, приведенные в настоящем стандарте, также могут относиться и к деталям, сечение которых не является окружностью |
| Код ОСК | 17.040.10 |
 Документ был принят в МГС 2013-11-14 и начал действовать 2015-07-01. Дата последнего издания 2014-10-02. Сейчас документ действует в таких странах: Республика Беларусь;Республика Казахстан;Кыргызская Республика;Российская Федерация;Республика Таджикистан;Украина.
Документ был принят в МГС 2013-11-14 и начал действовать 2015-07-01. Дата последнего издания 2014-10-02. Сейчас документ действует в таких странах: Республика Беларусь;Республика Казахстан;Кыргызская Республика;Российская Федерация;Республика Таджикистан;Украина.ГОСТы которые могут вас заинтересовать
Список ГОСТов
ГОСТ 2689-54. Допуски и посадки размеров свыше 500…
2598.00р.
ГОСТ 3047-66. Допуски и посадки размеров менее 1 м…
2028.00р.
ГОСТ 7713-62. Допуски и посадки.
 Основные определе…
Основные определе…2028.00р.
ГОСТ 8809-71. Допуски и посадки размеров менее 0,1…
1458.00р.
ГОСТ 11472-69. Допуски и посадки. Классы точности …
1458.00р.
ГОСТ 11710-66. Допуски и посадки деталей из пластм…
2028.00р.
ГОСТ 25307-82.
 Основные нормы взаимозаменяемости. …
Основные нормы взаимозаменяемости. …2598.00р.
ГОСТ 25346-89. Основные нормы взаимозаменяемости. …
2598.00р.
Биоаналоги соответствуют стандартам взаимозаменяемости снастей
Опубликовано:
Фелиза Мирасол
BioPharm International , BioPharm International-04-05-2020, выпуск 4 с: 28–32
Демонстрация взаимозаменяемости может обеспечить взаимозаменяемость биосимиляров на уровне аптеки.
В мае 2019 г. FDA опубликовало окончательное руководство (1) по демонстрации взаимозаменяемости биосимиляров и рекомендовало, какие аналитические оценки и данные должны быть включены, чтобы биоаналог мог считаться заменой назначенного оригинального биологического препарата. В отличие от непатентованных версий низкомолекулярных инновационных препаратов, биоаналоги нельзя просто заменить фирменными биологическими препаратами.
В отличие от непатентованных версий низкомолекулярных инновационных препаратов, биоаналоги нельзя просто заменить фирменными биологическими препаратами.
Вариабельность играет роль
Непатентованная версия низкомолекулярного лекарственного средства должна быть показана как химически идентичная своему предшественнику, но термин «идентичный» не может использоваться в обсуждении биологических препаратов по той простой причине, что существует вариабельность среди всех биологических препаратов, включая эталонные продукты, отмечает СангДжун Ли, старший исполнительный вице-президент Celltrion. «Эти присущие изменчивости основаны на таких факторах, как гетерогенное производство белка из клеточной линии живого организма и изменения в производственных процессах. Компании, производящие биосимиляры, должны измерять степень вариаций в разных партиях оригинального биологического препарата, чтобы рассматривать эти вариации как приемлемые границы для биоаналога», — говорит Ли.
Поскольку биоаналоги, в отличие от низкомолекулярных препаратов, производятся в живых клеточных линиях с использованием процессов, которые не могут быть точно воспроизведены от одного производителя к другому, производители биосимиляров должны продемонстрировать, что биоаналог очень похож на существующий FDA и не имеет клинически значимых отличий от него. — одобренный эталонный продукт с точки зрения безопасности и эффективности, — добавляет Леа Кристл, исполнительный директор по глобальному регулированию и политике исследований и разработок, Amgen.
— одобренный эталонный продукт с точки зрения безопасности и эффективности, — добавляет Леа Кристл, исполнительный директор по глобальному регулированию и политике исследований и разработок, Amgen.
«Для поддержки определения взаимозаменяемости необходимы дополнительные доказательства, — продолжает Кристл, — если FDA определит, что биоаналог соответствует стандарту взаимозаменяемости, этот биоаналог может быть заменен в аптеке в соответствии с законами штата об аптеках».
«Рынок биосимиляров в США отличается от других жестко регулируемых рынков, таких как Европа, Канада, Япония или Австралия, тем, что путь регулирования в Соединенных Штатах включает определение «взаимозаменяемости» между эталонным продуктом и биоаналогом. обозначение, которое позволяет фармацевтам заменять референтное лекарство биоаналогом без предварительного получения разрешения от лечащего врача. Без этого обозначения фармацевтам сначала необходимо связаться и получить разрешение от лечащего врача, прежде чем они заменят оригинальный биологический препарат биоаналогом», — говорит Гилель П. Коэн, доктор философии, исполнительный директор по научным вопросам Sandoz, компании Novartis.
Коэн, доктор философии, исполнительный директор по научным вопросам Sandoz, компании Novartis.
Однако биоаналог, одобренный FDA, не обязательно нуждается в обозначении взаимозаменяемости для одобрения оригинального биологического препарата по всем показаниям, отмечает Ноэль Санстром, основатель и генеральный директор NeuClone, австралийской биофармацевтической компании клинической стадии. «Например, хотя ни один из пяти биоаналогов Герцептина (трастузумаб), одобренных FDA, не обозначен как «взаимозаменяемый», все они одобрены для тех же показаний, что и оригинальный препарат. Взаимозаменяемость относится только к практике фармацевта, заменяющего оригинальный биологический препарат биоаналогом», — подчеркивает Санстром.
«Основная причина, по которой биоаналог не предназначен для «простой» замены, как и низкомолекулярный дженерик, связана с изменчивой структурой биологических препаратов, которая может влиять на их активность и период полувыведения из крови у пациентов. «, — говорит Санстром. «Для низкомолекулярных препаратов точная атомная структура исходного продукта четко определена и, как ожидается, не изменится. Таким образом, после истечения срока действия патента на оригинальное низкомолекулярное лекарство другой производитель может производить непатентованную альтернативу, которая является точной копией оригинального препарата и может строго контролироваться вплоть до атомарного уровня».
«Для низкомолекулярных препаратов точная атомная структура исходного продукта четко определена и, как ожидается, не изменится. Таким образом, после истечения срока действия патента на оригинальное низкомолекулярное лекарство другой производитель может производить непатентованную альтернативу, которая является точной копией оригинального препарата и может строго контролироваться вплоть до атомарного уровня».
«Однако с биологическими препаратами такая точная репликация не может быть достигнута, потому что все биологические препараты — будь то оригинал или биоаналог — варьируются от одной партии к другой. Эта неотъемлемая вариация является результатом использования живых клеток, используемых для производства биологических препаратов, которые представляют собой гетерогенные смеси различных молекул с различными посттрансляционными модификациями (ПТМ). Таким образом, биоаналоги могут быть «очень похожи» на оригинальный биологический препарат, который сам демонстрирует изменчивость от партии к партии», — поясняет Санстром.
Вариации PTM белка, включая профиль гликозилирования молекулы и образование дисульфидов, зависят от клеточной линии, говорит Мэтт МакГанн, менеджер по полевым приложениям и маркетингу в RedShiftBio, поставщике аналитического оборудования из Берлингтона, Массачусетс.
«Многие инновационные фармацевтические компании пошли на многое, чтобы разработать уникальные клеточные линии для производства биотерапевтического продукта, тем самым ограничивая возможность простого копирования лекарственного вещества», — говорит МакГанн.
«Процессы, связанные с разработкой биоаналога, приводят к изменению структуры белка от первичной до четвертичной», — продолжает МакГанн. «Новые рекомендации FDA по взаимозаменяемости биосимиляров подчеркивают эти различия, рекомендуя устанавливать биоподобие не только на основе фармакологических свойств лекарственного вещества, но и клинических данных, таких как клинические конечные точки и взаимозаменяемость. Демонстрация такого же или лучшего исхода для пациента считается необходимым доказательством для обоснования лицензирования биоаналога препарата, которое описано в совокупности доказательств FDA».
Уточнение взаимозаменяемости
Для практических целей разъяснение того, что означает «взаимозаменяемость», полезно при разработке биосимиляров. «Взаимозаменяемый» означает «биоаналог плюс», когда продукт должен сначала соответствовать требованиям, предъявляемым к биоаналогу, отмечает Дженнифер Мэллори, партнер Nelson Mullins Riley & Scarborough, юридической фирмы из Колумбии, Южная Каролина. После этого производитель биосимиляров должен соответствовать повышенным требованиям к оценке и тестированию.
«Они предназначены для обеспечения того, чтобы биоаналог давал тот же клинический результат, что и одобренный биологический препарат у любого пациента, и чтобы риски безопасности и эффективности, возникающие при переключении между одобренным биологическим препаратом и биоаналогом, не превышали риск использования только утвержденного биологическое, — говорит Мэллори.
Дополнительные препятствия предназначены для обеспечения возможности замены эталонного биологического препарата биоаналогом без необходимости вмешательства поставщика медицинских услуг, который прописал исходный эталонный биологический препарат, добавляет Мэллори.
В соответствии со стандартом FDA производитель предлагаемого взаимозаменяемого биоаналога должен показать, что продукт очень похож на референтный продукт и не имеет клинически значимых отличий от него, заявляет Christl. Кристл также указывает, что биоподобие не эквивалентно взаимозаменяемости. Последний предназначен для поддержки автоматического замещения на уровне аптеки и должен быть установлен в каждом конкретном случае, также говорит Кристл.
«Важно отметить, что биоаналог без обозначения взаимозаменяемости не уступает биоаналогу с обозначением», — отмечает Кристл. «Вместо этого обозначение взаимозаменяемости отражает определение FDA о том, что биоаналог может быть заменен референтным продуктом в аптеке без согласия врача. Это определение частично основано на дополнительных данных, показывающих, что пациенты могут переключаться между эталонным продуктом и биоаналогом без дополнительного риска».
Основываясь на собственном внутреннем анализе, Amgen полагает, что только около 10% биологических продуктов (за исключением инсулина и человеческого гормона роста) потеряют к 2023 году исключительные права в США. Эти продукты отпускаются в розничных аптеках, где возможна автоматическая замена, отмечает Кристл. «Мы видим, что многие биоаналоги вносят свой вклад в конкуренцию и получают широкое распространение в США, хотя FDA еще не определило ни один биоаналог как взаимозаменяемый».
Эти продукты отпускаются в розничных аптеках, где возможна автоматическая замена, отмечает Кристл. «Мы видим, что многие биоаналоги вносят свой вклад в конкуренцию и получают широкое распространение в США, хотя FDA еще не определило ни один биоаналог как взаимозаменяемый».
Кристл также заявляет, что руководство FDA по взаимозаменяемости по существу не касается аналитических испытаний или аналитических стандартов, помимо того, что агентство уже рассмотрело в более раннем руководстве по демонстрации биоподобия. «Что касается клинических испытаний, в руководстве FDA по взаимозаменяемости говорится, что спонсоры, ищущие обозначение взаимозаменяемости для продукта, предназначенного для использования одним человеком более одного раза, как правило, должны проводить исследования перехода, в которых пациенты начинают с оригинального продукта и случайным образом распределяются. перейти на биоаналог или продолжить использование оригинального продукта. Согласно руководству, ожидается, что группа «переключения» будет включать как минимум два переключения между оригинальным и биоаналогичным продуктом и как минимум два контакта с каждым продуктом», — говорит Кристл.
«Amgen считает, что текущие стандарты FDA для демонстрации взаимозаменяемости являются научно обоснованными», — добавляет она.
«Для биоаналога очень важно продемонстрировать «взаимозаменяемость», поскольку, в отличие от дженериков, биоаналог может быть назначен врачом в соответствии с действующим законодательством. Несмотря на использование биосимиляров на протяжении многих лет, были сомнения в их назначении из-за потенциальных рисков, таких как снижение эффективности или ожидание иммуногенности после перехода на биоаналог», — говорит Ли. Однако Ли отмечает, что обозначение взаимозаменяемости на основе результатов эффективности многократного переключения будет побуждать врачей и пациентов чаще использовать биоаналоги. Кроме того, статус взаимозаменяемости в конечном итоге позволяет отпускать любой продукт из числа доступных взаимозаменяемых продуктов на уровне аптеки, что, как ожидается, ускорит доступ к биоаналогам.
Взаимозаменяемое обозначение в США требует дополнительных клинических данных помимо предоставленных для установления биоподобия, указывает Коэн. «Это не более высокий стандарт качества, потому что взаимозаменяемый биологический препарат и биоаналог по определению соответствуют эталонному продукту по безопасности, чистоте, активности и эффективности.
«Это не более высокий стандарт качества, потому что взаимозаменяемый биологический препарат и биоаналог по определению соответствуют эталонному продукту по безопасности, чистоте, активности и эффективности.
Важен ли статус взаимозаменяемости?
Важность взаимозаменяемости особенно актуальна для биологических препаратов, выдаваемых фармацевтами пациентам с хроническими заболеваниями, поскольку эти пациенты нуждаются в лечении в течение длительного периода времени, отмечает Санстром.
Однако Санстром объясняет, что если лечение проводится только в течение короткого периода времени, как, например, при лечении многих онкологических биологических препаратов, пациент может получать меньше доз, тем самым уменьшая возможность переключения между оригинальным препаратом и биоаналогом. Если врач также назначает биопрепарат (т. е. фармацевт не принимает решения), он имеет полный контроль над тем, вводится ли оригинальный препарат или биоаналог, без возможности замены на уровне аптеки, что снижает статус взаимозаменяемости в подобных случаях. значительный.
значительный.
«Обратите внимание, что по состоянию на 12 марта 2020 г. ни один из 26 биоаналогов, одобренных Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA), не был признан взаимозаменяемым», — отмечает Санстром.
Кроме того, ведутся споры о необходимости обозначения взаимозаменяемости FDA вообще. «Взаимозаменяемость является дополнительным атрибутом биоаналога, который достигается за счет значительных инвестиций в дизайн клинических испытаний, который включает в себя смену оригинального препарата на биоаналог и обратно в исследованиях с большим числом пациентов. Это не означает, что взаимозаменяемый биоаналог каким-либо образом превосходит биоаналог, не имеющий такого обозначения», — заявляет Санстром.
Коэн придерживается аналогичного мнения, говоря, что с научной точки зрения биоаналог разрабатывается так, чтобы соответствовать эталонному лекарству по этим характеристикам. «Поэтому наша позиция заключается в том, что утвержденный биоаналог следует считать взаимозаменяемым с референтным лекарством, а это означает, что пациент может ожидать эквивалентного результата лечения», — добавляет Коэн.
«На сегодняшний день существует 26 биоаналогов, одобренных FDA, но только 16 продаются в США [на момент публикации]. Для сравнения, Европейское агентство по лекарственным средствам (EMA) одобрило 64 биоаналога, 59из которых доступны для использования в Европе. Хотя нас воодушевляет то, что системы здравоохранения в США все чаще внедряют биоаналоги в свою клиническую практику, все еще существуют препятствия, которые необходимо устранить для поддержки рынка биоаналогов, который может обеспечить успех, подобный тому, что мы видели в Европе». констатирует Коэн.
Одним из препятствий являются проблемы, связанные с процессом утверждения FDA взаимозаменяемости, в частности тот факт, что для получения взаимозаменяемости спонсоры должны провести обширное сравнительное фармакокинетическое (ФК) исследование с использованием нескольких переключений с последующим продолжительным периодом наблюдения. «Этот критерий не обязателен для фармакокинетических исследований оригинальных биологических препаратов, когда в производственный процесс вносятся изменения, которые приводят к структурным изменениям самого биологического препарата. Дополнительные затраты на исследования и затраты на то, чтобы биосимилярный препарат считался взаимозаменяемым, значительны и могут удерживать производителей биосимиляров от стремления к взаимозаменяемости», — подчеркивает Коэн.
Дополнительные затраты на исследования и затраты на то, чтобы биосимилярный препарат считался взаимозаменяемым, значительны и могут удерживать производителей биосимиляров от стремления к взаимозаменяемости», — подчеркивает Коэн.
Таким образом, значение взаимозаменяемого атрибута находится в автоматической подстановке на уровне аптеки. «В Европе нет эквивалентного обозначения от Европейского агентства по лекарственным средствам (EMA), и переключение между оригинальными препаратами и биоаналогами является медицинским вопросом, как правило, на усмотрение лечащего врача, а не фармацевта. Как утверждают Ebbers & Schellekens (2019), биоаналоги должны быть взаимозаменяемыми по умолчанию, а не должны соответствовать дополнительным требованиям, чтобы быть обозначенными как таковые: «нет причин сомневаться в том, что биоаналоги взаимозаменяемы и что риск повышения иммуногенности при переходе на биоаналог не более чем переключение между двумя партиями любого биологического препарата. Мы утверждаем, что по умолчанию биоаналоги должны быть взаимозаменяемыми, если нет убедительных доказательств обратного». Санстром цитирует (2).
Санстром цитирует (2).
Аналитическое руководство
Когда дело доходит до аналитических рекомендаций в руководстве по взаимозаменяемости FDA, ключевая фраза, которую следует принять к сведению, содержится в разделе V.A.1.: «Достижения в области аналитики могут позволить получить расширенную аналитическую характеристику, которая повлияет на объем других данные и информация, необходимые для демонстрации взаимозаменяемости…» (1).
МакГанн отмечает, что это заявление подчеркивает тот факт, что усовершенствование аналитических методов для характеристики биотерапевтических средств может существенно повлиять на совокупность доказательств, необходимых для поддержки утверждений о биоподобии. «Улучшения в возможностях приборов, таких как чувствительность, разрешение или точность, которые могут более четко продемонстрировать биологическое сходство, могут уменьшить бремя доказывания, необходимого для демонстрации как биоподобия, так и взаимозаменяемости», — говорит МакГанн.
Усовершенствованные аналитические инструменты (например, AQS3pro, RedShiftBio) могут обеспечить 30-кратное повышение чувствительности и возможность измерения вторичной структуры в условиях рецептуры. Последнее измерение ранее было невозможно, и оно может предоставить доказательства биоподобия, что снижает бремя доказывания в других областях, говорит МакГанн.
«По этой причине разработчики биосимиляров должны убедиться, что их аналитические инструменты находятся на переднем крае технологий. Как компания, они должны постоянно пересматривать свои возможности, чтобы оставаться в авангарде технологий по сравнению с рынком», — добавляет МакГанн.
«В последние годы FDA заняло менее предписывающую позицию, когда речь идет о том, какие типы аналитических характеристик следует использовать, предпочитая перекладывать ответственность на фармацевтическую компанию, чтобы обосновать, почему измерение не требуется, а не запрашивать конкретные испытания, которые необходимо выполнить. Это изменение можно объяснить быстрым изменением технологий в последние годы», — объясняет МакГанн.
Это изменение можно объяснить быстрым изменением технологий в последние годы», — объясняет МакГанн.
Celltrion поддерживает эту точку зрения. Ли говорит, что вместо предоставления каких-либо конкретных аналитических рекомендаций, которые могут реализовать разработчики биосимиляров, в новом руководстве объясняется позиция агентства, в котором обсуждается планируемый подход к разработке. Этот подход включает любое предлагаемое обоснование сокращения объема информации, необходимой для подтверждения взаимозаменяемости посредством расширенной аналитической характеристики. «Наша компания планирует обсудить разумные подходы к каждому продукту с регулирующим органом, чтобы уменьшить нагрузку на клинические испытания», — заявляет Ли.
Руководство содержит примеры, которые спонсоры могут использовать в качестве контрольных показателей для оценки подходов, которые они планируют использовать для демонстрации взаимозаменяемости и разработки плана разработки своего продукта, говорит Мэллори. Мэллори также отмечает, что желательно указать подходы, выделенные в руководящем документе, которые спонсор уже использовал в своей работе по разработке и ранее представил FDA в связи с процессом утверждения биосимиляров. «Точно так же спонсорам было бы полезно определить, какие подходы в руководящем документе кажутся неприменимыми (и почему), а также любые потенциально доступные короткие пути. Часть этого анализа в идеале должна быть проведена еще до предварительных обсуждений с FDA, чтобы спонсор мог быть уверен в конкретных целях и задачах, которые FDA дало зеленый свет или начало рассматривать», — подчеркивает она.
Мэллори также отмечает, что желательно указать подходы, выделенные в руководящем документе, которые спонсор уже использовал в своей работе по разработке и ранее представил FDA в связи с процессом утверждения биосимиляров. «Точно так же спонсорам было бы полезно определить, какие подходы в руководящем документе кажутся неприменимыми (и почему), а также любые потенциально доступные короткие пути. Часть этого анализа в идеале должна быть проведена еще до предварительных обсуждений с FDA, чтобы спонсор мог быть уверен в конкретных целях и задачах, которые FDA дало зеленый свет или начало рассматривать», — подчеркивает она.
Важной темой руководства, подчеркивает Мэллори, является то, что FDA не предлагает универсального подхода для демонстрации взаимозаменяемости. Скорее, агентство использовало руководство, чтобы подтвердить, что демонстрация взаимозаменяемости сложна, как и сложные молекулы. «Таким образом, FDA отметило в руководстве, что «данные и информацию, необходимые для демонстрации взаимозаменяемости, необходимо рассматривать в каждом конкретном случае», — говорит Мэллори.
Учитывая точку зрения FDA, полезно, чтобы в руководстве неоднократно подчеркивалось, что спонсорам рекомендуется обращаться в агентство для проведения предварительных обсуждений и планирования встреч. «Кроме того, как указано в уведомлении Федерального реестра относительно руководства, FDA не предполагает, что это будет его последнее слово в отношении взаимозаменяемости. Действительно, в феврале 2020 г. FDA выпустило дополнительный проект руководства под названием «Биоаналоги и взаимозаменяемые биоаналоги: лицензирование не для всех условий использования, для которых был лицензирован эталонный продукт» (3). Мы можем ожидать предстоящих руководств агентства и других действий, которые, как мы надеемся, дадут больше информации разработчикам биоаналогов и взаимозаменяемых биоаналогов», — заключает Мэллори.
«Аналитические процессы и инструменты, используемые при разработке биосимиляров, очень похожи на те, которые используются при разработке оригинальных биологических продуктов. Основное различие заключается в том, как эти инструменты используются в итеративном процессе разработки биосимиляров. Хотя методы и инструменты технически сложны (а значит, не просты), их можно довольно просто использовать для разработки биоаналогов, которые соответствуют критериям «очень похожи без клинически значимых различий», — добавляет Кристл.
Основное различие заключается в том, как эти инструменты используются в итеративном процессе разработки биосимиляров. Хотя методы и инструменты технически сложны (а значит, не просты), их можно довольно просто использовать для разработки биоаналогов, которые соответствуют критериям «очень похожи без клинически значимых различий», — добавляет Кристл.
Ссылки
1. FDA, Руководство для промышленности, Рекомендации по демонстрации взаимозаменяемости с эталонным продуктом Руководство для отрасли (CDER, CBER, май 2019 г.).
2. Х.К. Эбберс и Х. Шеллекенс, Drug Discovery Today , 24 (10) 1963–1967 (2019).
3. FDA, Проект отраслевого руководства, биоаналоги и взаимозаменяемые биоаналоги: лицензирование менее чем для всех условий использования, для которых эталонный продукт был лицензирован Промышленное руководство (CDER, CBER, февраль 2020 г.).
Информация о статье
BioPharm International
Vol. 33, No. 4
4
April 2020
Страницы: 28–32
Citation
При ссылке на эту статью просьба ссылаться на нее как на F. Mirasol, «Biosimilars Tackle Interchangeability Standards», BioPharm International 33 (4) 2020.
Загрузить Выпуск: BioPharm International-04-01-2020
Связанный контент:
Качество/GMPsBiosimilars and BiobettersАналитические методыБиологические препараты в разработкеНормативные заявки и заявкиBioPharm International-04-01-20200005
Связанная статья >>>
FDA завершает работу над руководством по взаимозаменяемости биоаналогов, повторяет индивидуальный подход к требованиям к данным
10 мая 2019 г. -производителям больше ясности в отношении того, как продемонстрировать, что предлагаемый биоаналог соответствует установленному законом стандарту взаимозаменяемости в соответствии с Законом об общественном здравоохранении (Закон о PHS или Закон). Согласно Закону, взаимозаменяемый биоаналог может быть заменен оригинальным биологическим продуктом без участия врача, назначающего лекарство, аналогично тому, как непатентованные препараты обычно заменяются на патентованные препараты на уровне аптеки. Окончательное руководство, озаглавленное «Соображения по демонстрации взаимозаменяемости с эталонным продуктом», короче, чем черновая версия, выпущенная более двух лет назад в ответ на отзывы отрасли, но в целом соответствует исходным положениям политики, предложенным в черновике, с некоторыми примечательными исключения приведены ниже.
Согласно Закону, взаимозаменяемый биоаналог может быть заменен оригинальным биологическим продуктом без участия врача, назначающего лекарство, аналогично тому, как непатентованные препараты обычно заменяются на патентованные препараты на уровне аптеки. Окончательное руководство, озаглавленное «Соображения по демонстрации взаимозаменяемости с эталонным продуктом», короче, чем черновая версия, выпущенная более двух лет назад в ответ на отзывы отрасли, но в целом соответствует исходным положениям политики, предложенным в черновике, с некоторыми примечательными исключения приведены ниже.
Введение к взаимозаменяемым биоаналогам
Нет требования, чтобы новый биоаналог демонстрировал себя как «взаимозаменяемый» по отношению к эталонному биологическому продукту, то есть оригинальный биологический продукт, на который спонсор биоаналога стремится полагаться в своих собственных маркетинговое одобрение. Закон предусматривает две категории лицензированных последующих биологических препаратов: биоаналог и взаимозаменяемый биоаналог, и данное новое Заключительное руководство не меняет эту структуру. И хотя FDA не лицензировало взаимозаменяемые биоаналоги с момента принятия Закона о ценовой конкуренции и инновациях в области биологических препаратов (BPCIA), который внес поправки в Закон о PHS в 2010 году, чтобы создать законный путь для биоаналогов, по крайней мере 45 штатов приняли местные законы или правила в чтобы разрешить лицензированным специалистам в области здравоохранения заменять одобренные FDA взаимозаменяемые биоаналоги, если и когда они поступят на рынок (см. наш последний список этих законов штата здесь).
И хотя FDA не лицензировало взаимозаменяемые биоаналоги с момента принятия Закона о ценовой конкуренции и инновациях в области биологических препаратов (BPCIA), который внес поправки в Закон о PHS в 2010 году, чтобы создать законный путь для биоаналогов, по крайней мере 45 штатов приняли местные законы или правила в чтобы разрешить лицензированным специалистам в области здравоохранения заменять одобренные FDA взаимозаменяемые биоаналоги, если и когда они поступят на рынок (см. наш последний список этих законов штата здесь).
Так что же такое взаимозаменяемый биоаналог? В дополнение к соответствию различным критериям «биоаналогичности» или высокой степени сходства с эталонным биологическим препаратом, как FDA объяснило эту концепцию в других отраслевых руководствах, Закон PHS требует, чтобы продукт соответствовал следующим двум критериям, прежде чем он может быть лицензирован как взаимозаменяемо:
- Можно ожидать, что биоаналог даст тот же клинический результат, что и эталонный биологический препарат у любого данного пациента; и
- Для продукта, который будет вводиться пациентам более одного раза, «риск с точки зрения безопасности или снижения эффективности чередования или переключения между использованием [биоаналога] и эталонного продукта не превышает риск использования эталонного продукта без такого чередования или переключения».

Биологические препараты остаются одними из самых дорогих продуктов на рынке из-за сложности их получения из живых клеток и того факта, что такие продукты используются для лечения серьезных хронических заболеваний, таких как рак, рассеянный склероз и аутоиммунные заболевания. Конгресс ввел эти повышенные критерии взаимозаменяемости в попытке усилить конкуренцию и стимулировать разработку более дешевых альтернатив биологическим продуктам, которые в долгосрочной перспективе сократят расходы для пациентов, правительств и страховых компаний, в то же время защищая безопасность пациентов и гарантируя, что медицинские и научные вопросы учитываются при разработке продукта и предпродажном нормативном рассмотрении. На сегодняшний день FDA одобрило 19биоаналогов, но, как отмечалось ранее, ни один из них не был одобрен как взаимозаменяемый с эталонным биологическим препаратом. Требовательные критерии FDA для этого обозначения взаимозаменяемости и отсутствие ясности в отношении того, как достичь или продемонстрировать, что критерии были соблюдены, называются факторами, способствующими минимальному прогрессу в этой области за последние несколько лет.
Окончательная политика взаимозаменяемости более гибкая, чем первые предложения FDA
Долгожданное Окончательное руководство содержит рекомендации для производителей биосимиляров и обеспечивает большую гибкость в отношении дизайна исследований, необходимых для демонстрации того, что критерии взаимозаменяемости были созданы. В заявлении, сделанном в тот же день, исполняющий обязанности комиссара FDA Нед Шарплесс резюмировал новый документ следующим образом: «Сегодняшнее окончательное руководство дает обзор важных научных соображений при демонстрации взаимозаменяемости с эталонным продуктом и объясняет научные рекомендации для применения или дополнения к предлагаемый взаимозаменяемый продукт».
В частности, окончательное руководство FDA по взаимозаменяемости касается следующих важных научных тем, по которым спонсоры биосимиляров добивались большей ясности:
- Какие данные и информация необходимы для демонстрации взаимозаменяемости;
- Рекомендации по планированию и анализу исследования или исследований по переключению для демонстрации взаимозаменяемости;
- Соображения относительно продукта сравнения в исследовании или исследованиях переключения; и
- Соображения по разработке презентаций, систем укупорки контейнеров и составных частей устройств доставки для предлагаемых взаимозаменяемых продуктов (последняя тема рассматривается в сокращенной форме, поскольку они будут оцениваться в каждом конкретном случае в зависимости от продукта) ).

По сравнению с предложенной политикой взаимозаменяемости, опубликованной в январе 2017 г., FDA не изменило свою первоначальную рекомендацию о том, что спонсор биосимиляра должен представлять данные и информацию, чтобы показать, что предлагаемый взаимозаменяемый продукт «можно ожидать того же клинического результата, что и эталонный препарат». биологический» (критерий № 1, см. выше) для всех лицензионных условий использования эталонного продукта. Некоторые комментаторы проекта руководства просили пересмотреть его, чтобы учесть тот факт, что биоаналог — так же, как и непатентованный препарат — может быть одобрен FDA по меньшему количеству показаний, по которым может быть одобрен референтный продукт, из-за защиты патента или эксклюзивности. по подмножеству показаний.
Тем не менее, одно из наиболее значительных изменений в окончательных рекомендациях FDA является долгожданным для все еще зарождающейся индустрии биоаналогов США. Агентство отошло от формулировки, указывающей, что только эталонные биологические препараты, лицензированные FDA, могут использоваться для проведения «переходных исследований», которые будут необходимы в большинстве случаев для соответствия стандарту, установленному критерием взаимозаменяемости № 2 (см. выше). Исследования перехода должны проводиться при одном или нескольких условиях использования, не обязательно при всех показаниях, для которых лицензирован референтный препарат, и Агентство рекомендует использовать условие, «которое будет адекватно чувствительным для оценки риска чередования или переключения» и что будет поддерживать научное обоснование экстраполяции данных на другие условия использования. Кроме того, спонсору биоаналога по-прежнему необходимо будет установить научный мост между препаратом сравнения, не лицензированным в США, и версией, лицензированной FDA, чтобы обосновать перед Агентством, почему исследование перехода является научно обоснованным и обоснованным:
выше). Исследования перехода должны проводиться при одном или нескольких условиях использования, не обязательно при всех показаниях, для которых лицензирован референтный препарат, и Агентство рекомендует использовать условие, «которое будет адекватно чувствительным для оценки риска чередования или переключения» и что будет поддерживать научное обоснование экстраполяции данных на другие условия использования. Кроме того, спонсору биоаналога по-прежнему необходимо будет установить научный мост между препаратом сравнения, не лицензированным в США, и версией, лицензированной FDA, чтобы обосновать перед Агентством, почему исследование перехода является научно обоснованным и обоснованным:
Если спонсор хочет использовать данные, полученные в результате исследования перехода или исследований, сравнивающих предлагаемый взаимозаменяемый продукт с продуктом сравнения, не лицензированным в США, как часть демонстрации того, что предлагаемый взаимозаменяемый продукт соответствует стандарту, описанному в … Законе о PHS, Спонсор должен предоставить адекватные данные и информацию для установления «моста» между препаратом сравнения, не лицензированным в США, и эталонным продуктом, лицензированным в США, и тем самым обосновать релевантность данных, полученных с использованием препарата сравнения, не лицензированного в США, для оценки соответствия требованиям.
[Закона] были соблюдены.
Несмотря на то, что параметры дизайна для этих исследований перехода являются гибкими, и FDA рекомендует спонсорам консультироваться со своим персоналом по биоаналогам по конкретным соображениям дизайна, таким как первичные конечные точки и статистический анализ, Агентство по-прежнему подходит к этому критерию взаимозаменяемости с позиции, что исследования переключения всегда будут требуется для проведения необходимых научных исследований по критерию №2. Таким образом, любой спонсор биоаналога, который не считает, что исследование перехода необходимо, вероятно, будет нести тяжелое бремя убеждения FDA отказаться от таких данных, когда биоаналог предназначен для введения пациентам более одного раза. По этому поводу в Заключительном руководстве говорится: «Если спонсор предлагаемого взаимозаменяемого продукта считает, что данные исследования перехода не нужны, FDA ожидает, что спонсор предоставит обоснование того, что такие данные не нужны, как часть демонстрации эффективности. взаимозаменяемость». Далее следует отметить, однако, что дизайн исследований перехода может зависеть от ожиданий спонсора относительно того, как предлагаемый взаимозаменяемый биоаналог может быть использован в клинической практике.
взаимозаменяемость». Далее следует отметить, однако, что дизайн исследований перехода может зависеть от ожиданий спонсора относительно того, как предлагаемый взаимозаменяемый биоаналог может быть использован в клинической практике.
Наконец, д-р Шарплесс указал в своем заявлении, сопровождающем окончательное руководство, что обозначение взаимозаменяемости направлено на повышение уверенности пациентов и врачей в безопасности и эффективности биоаналогов, что является одной из целей Плана действий по биоаналогам, опубликованного в середине 2018 года. Точно так же появление взаимозаменяемых биоаналогов на рынке США может помочь прояснить любую путаницу или разногласия между сообществами плательщиков и поставщиков медицинских услуг, когда речь идет о надлежащем использовании и покрытии этих продуктов.
Взаимозаменяемость в Статуте не включает исключений – но создаст ли FDA их для препаратов инсулина?
Следующим пунктом повестки дня FDA является рассмотрение вопроса об утверждении продуктов инсулина в качестве биоаналогов после того, как в марте 2020 года эти и некоторые другие лекарственные препараты перейдут из «пути одобрения лекарств» в «путь одобрения биологических препаратов», как это предписано съезд в БНКПА. Действительно, независимо от того, был ли выпуск Руководства по взаимозаменяемости от 10 мая намеренно приурочен к этому событию, Агентство созвало открытое собрание 14 мая 2019 г.услышать мнения заинтересованных сторон о разработке биоаналогов и взаимозаменяемых продуктов инсулина.
Действительно, независимо от того, был ли выпуск Руководства по взаимозаменяемости от 10 мая намеренно приурочен к этому событию, Агентство созвало открытое собрание 14 мая 2019 г.услышать мнения заинтересованных сторон о разработке биоаналогов и взаимозаменяемых продуктов инсулина.
Одно из мнений, высказанных во время встречи несколькими докладчиками, заключалось в том, что FDA не должно требовать переключения исследований до утверждения инсулина как взаимозаменяемого в соответствии с BPCIA, учитывая историю клинического опыта с этими продуктами и отсутствие опасений по поводу иммуногенности этого небольшого, простой белковый продукт. (Как выразился один публичный докладчик: «Инсулин — это не Хумира» — ссылка на тот факт, что продукты моноклональных антител намного крупнее и их значительно сложнее охарактеризовать, чем эти белки). Хотя давление на рынок инсулина уникально, и обсуждение сложностей, вызванных переходом от лекарств к биологическим препаратам в марте 2020 года, выходит за рамки этого сообщения в блоге, мы отмечаем это слияние вопросов, потому что будет интересно увидеть, как Агентство отвечает на эти аргументы и какую политику она объявляет в отношении продуктов инсулина, в частности.